
Sickle Cell Disease
Questions, Traits, and Responses.

By Dr. Sharon K. West –
Though September is the month designated to heighten awareness of Sickle Cell Disease (SCD) and Sickle Cell Trait (SCT), we all should be aware every month that many children, adults, and families are affected by this genetic disease.
With the colder temperatures approaching, this is especially difficult because the stress of temperature extremes can trigger significant painful episodes in those with sickle cell disease, often resulting in hospitalization.
Here are some questions and responses about Sickle Cell.
Sharon, I have heard of Sickle Cell Disease, but what is it? Also, can I “catch it” from someone who has it?
Great questions. First of all, both the disease and the genetic trait are inherited. They are not contagious—meaning “Nope,” you cannot “catch it” from anyone by a mere touch or living in the same household, etc.
As a genetic trait, Sickle Cell comes from one or both parents.
If one parent has SCD and other parent does not (has normal red blood cells), each child will have SCT.
If one parent has SCD and the other has SCT, there is 50% chance of having a baby with either SCD or SCT with each pregnancy.
When both parents have SCT, there is a 25% chance (1 out of 4) of having a baby with SCD with each pregnancy.
What population does SCD/SCT mostly affect?
One of the latest statistics I have seen is that SCD affects approximately 100,000 Americans. Most are African American with a smaller percentage of Hispanics, Middle Eastern, or Asian Indian populations being affected.
Is there a test for SCD/SCT?
At birth, a blood test will alert the medical team if this infant has SCD or SCT. Neither condition occurs later in life: one either is or isn’t born with this!
Before mandatory testing for SCD at birth was established in the United States in 2006, many people were not tested, so many adults may not know if they have it or not. Folks with SCT—the genetic trait—generally have no symptoms. Folks with SCD, the disease itself, usually do exhibit signs and symptoms, which can be quite painful and are mostly referred to as a sickle cell crisis.
What are the symptoms of SCD?
Based on my experience in working with patients with sickle cell disease, the pain is so intense that hospitalization, with analgesics such as opioids over a significant period, is basically the only way to address it.
I have read about several other approaches that can ward off pain crisis. I recall the stories from African American males living with Sickle Cell Disease stating that they always knew when a crisis was starting because of the discomfort. They would drive directly to the hospital so that the medical intervention could delay or prevent the process from going into a full pain crisis. However, by the time the medical team in emergency departments would do their assessment to rule out “drug-seeking” behavior, or get past their judgement of a black male seeking medicine for pain, the patients would transition into a full-blown crisis.
These stories were and are horrendous. Sufferers have lived their lives with this disease to only have their voices silenced or dismissed due to stereotypes. I wish I could say this happened infrequently.
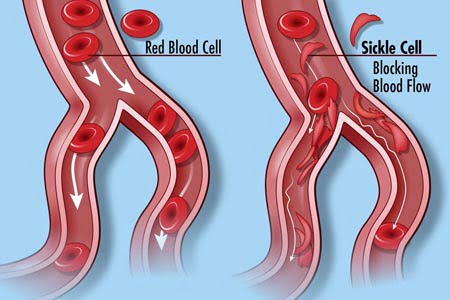
What do the RBCs do exactly? What causes the pain that folks with SCD have?
Red blood cells are the main substance of hemoglobin, which helps carry the oxygen from the air we breathe into our lungs and to all parts of the body. A normal red blood cell is disc-shaped (round), and normal RBCs last about 120 days before they are replaced by new ones. That round shape allows oxygen to flow freely through the small blood vessels in our body.
However, in a sickled cell, because of the presence of the “S hemoglobin” [S stands for sickle], the RBCs are shaped like a crescent moon—pointy on the ends. These red blood cells are stiff, so they don’t flow freely through the blood vessels, making it very difficult for oxygen to reach the vital organs. These cells only last approximately 16 days and are then replaced with the same type of affected cell.
When the body does not get the oxygen it needs from the RBCs, those major organs are soon damaged, and that is when the complications as a result of sickle cell disease begin.
What is effective in treating patients with Sickle Cell Disease?
Relieving pain and preventing complications is the general approach to treatment.
Having medical teams who are well versed on SCD is essential.
“Each one, reach one, to teach one” within and throughout the community on Sickle Cell Disease.
Blood transfusions are life-saving for those impacted by Sickle Cell Disease. The American Red Cross states that one of three black blood donors is a match for sickle cell patients. African Americans have unique structures on our red blood cells called antigens. These antigens are inherited, and many of those with sickle cell disease will require closely matched blood. In many cases, that closely matched blood comes from those of the same ethnicity.
In addition to blood transfusions, there are cases where blood and bone marrow transplants have demonstrated a cure. Also, the newer gene-splicing technology called Crispr is very promising as well.
The message about SCD and SCT
Sickle Cell Disease is real. Sickle Cell Trait is real.
As a person of color or of African Descent or Hispanic, Middle Eastern, or Asian Indian descent, do you know if you have SCT or SCD? Some churches, especially in the African American faith community, offer free sickle cell screening. Tried Stone Missionary Baptist Church in Asheville is one such location; they recently offered screenings at the Fall Wellness Fair. Ask your church if it offers screening for members and the community.
What if I screen positive?
Know your family medical history. If you have this diagnosis, make sure your family knows, because your children must be made aware if they themselves carry this trait.
What should medical personnel do?
First, do not dismiss the voices of those who have this diagnosis. Pain is what patients say it is, even walk-ins to your hospital or clinic. Therefore:
Leave your stereotypes at the door. Folks did not ask for this.
Be an advocate. Many times, folks wait until the very last minute to come to the hospital or clinic because of how poorly they were treated on previous occasions—or how they were made to feel lesser than or underserving of care, especially if insurance is an issue.
What should the community do?
Populations of color, donate blood. If you don’t carry SCT or SCD, you could be a crucial match for those in need. Call the American Red Cross and ask how you can be a donor.
Sororities and Fraternities of color, if you’re a member of the historical Divine Nine, coordinate an annual blood drive for sickle cell disease as a service project.
Care! As Dr. Maya Angelou wrote, “People don’t care how much you know until they know how much you care.”
Learn what you need as a member of a susceptible population to SCD and SCT—and show that you care!







